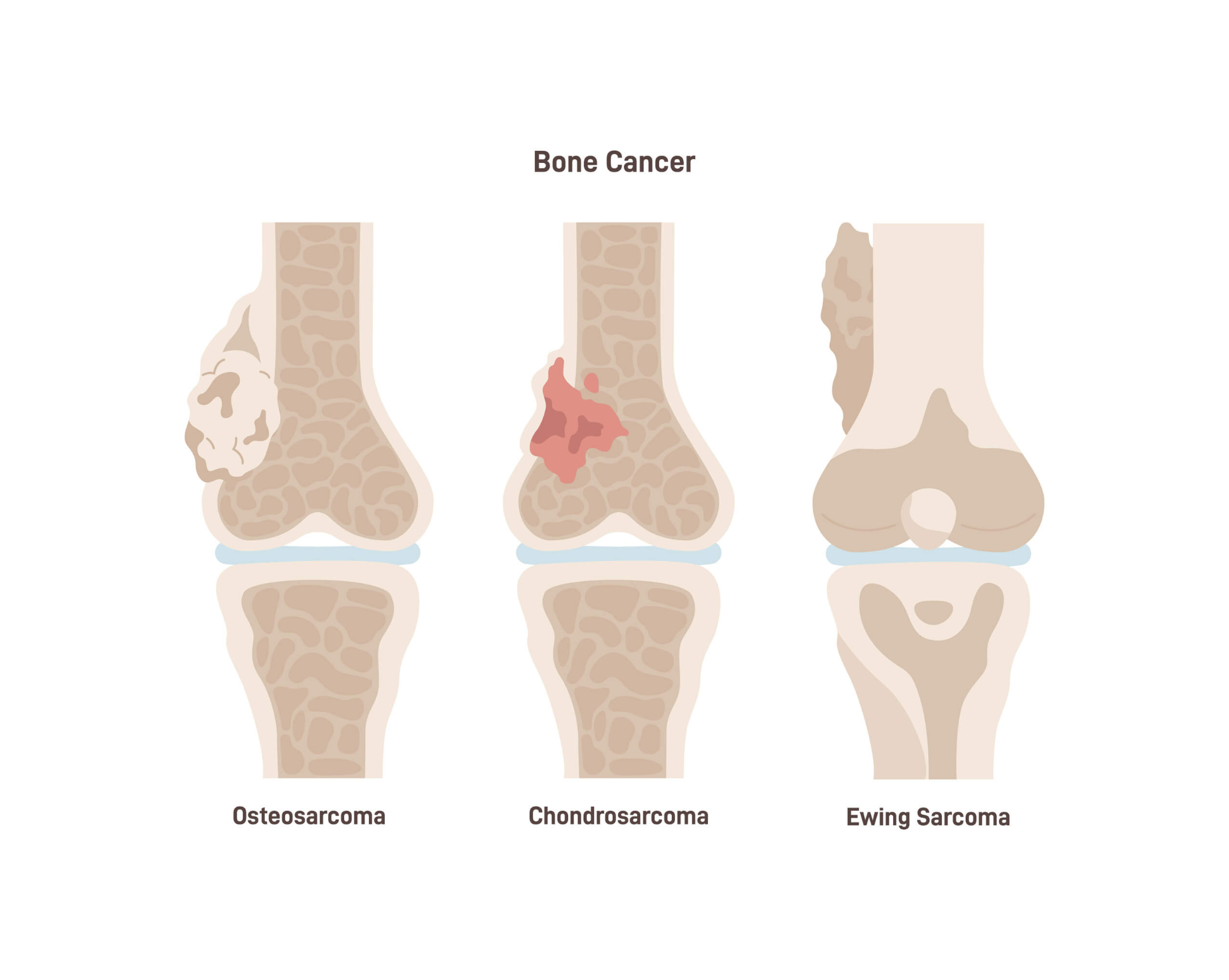
Das Ewing-Sarkom
Was ist das Ewing-Sarkom?
Das Ewing-Sarkom ist ein bösartiger Tumor, der meist vom Knochen ausgeht (Knochenkrebs). Seltener tritt er als reiner Weichgewebstumor auf. Seinen Namen hat der Tumor von dem Krebsforscher James Ewing. Der New Yorker hat die Tumorart erstmals im Jahr 1921 ausführlich untersucht und beschrieben.
Das Ewing-Sarkom ist eine rasch wachsende Krebsart, die Metastasen – Absiedlungen in anderen Körperregionen – bildet. Ohne eine vernünftige Therapie verläuft die Erkrankung tödlich.
Das Ewing-Sarkom kann an allen möglichen Stellen im Körper entstehen. Am häufigsten sind jedoch folgende Regionen betroffen:
- Becken
- Oberschenkel und Unterschenkel
- Rippen
- Schulterblatt
- Wirbelsäule
In etwa 20 % der Fälle tritt es als reiner Weichteiltumor – sogenanntes extraossäres Ewing-Sarkom – auf.
Im Zuge der Diagnosestellung werden bei rund 20 % der Patienten und Patientinnen bereits Metastasen festgestellt. Befallen sind meist die Lunge, andere Knochen oder das Knochenmark. Zusätzlich bestehen bei allen Betroffenen sogenannte Mikrometastasen – kleinste Tumorzellen, die sich über das Blut im Körper ausbreiten.
Mikrometastasen sind mit den herkömmlichen Untersuchungsverfahren nicht zu sehen, können aber vor Beginn einer Behandlung im Blut nachgewiesen werden. Aus diesem Grund wird das Ewing-Sarkom als Systemerkrankung bezeichnet. Die Krebserkrankung befällt nicht nur die Knochen oder das Weichgewebe, sondern den gesamten Körper.

Das Ewing-Sarkom ist selten
In Deutschland erkranken pro Jahr ca. 3 von 1 Millionen Kinder, Jugendliche und (junge) Erwachsene am Ewing-Sarkom. Jährlich werden hierzulande ca. 100 Patienten und Patientinnen mit diesem seltenen Knochenkrebs neu diagnostiziert.
Besonders häufig tritt die Erkrankung im Jugend- und jungen Erwachsenenalter auf. Jungen bzw. Männer sind etwas häufiger betroffen – 1,3:1 Geschlechterverhältnis. Gerade weil die Erkrankung so selten ist, sollte die Behandlung unbedingt in einem Sarkomzentrum stattfinden. Dort ist das behandelnde Team auf diese Erkrankung spezialisiert.
Ursachen des Ewing-Sarkoms
Bis heute ist nicht bekannt, warum manche Menschen das Ewing-Sarkom entwickeln. Auch die potenziellen Risikofaktoren, die den Krebs in seiner Entwicklung begünstigen, lassen sich nicht genau definieren. Auch wenn der seltene Knochenkrebs besonders häufig im zweiten Lebensjahrzent auftritt, können auch Säuglinge oder alte Menschen betroffen sein. Die ethnische Herkunft scheint dabei eine Rolle zu spielen. Die Inzidenz liegt bei der weißen oder kaukasischen Bevölkerung mit ca. 1,5/1.000.000 doppelt so hoch wie bei der asiatischen Bevölkerung– ca. 0,8/1.000.000. Bei Menschen afrikanischer Herkunft ist das Ewing-Sarkom dagegen nahezu unbekannt (Inzidenz ca. 0,2/1.000.000), diese Daten sind wissenschaftlich belegt und haben nichts mit Diskriminierung von Bevölkerungsgruppen gemein.
Selten tritt das Ewing-Sarkom im Rahmen von Tumorprädispositionssyndromen auf. Dabei handelt es sich um eine Gruppe von Krankheiten, bei denen aufgrund einer genetischen Veränderung (Mutation) ein höheres Krebsrisiko besteht.
Genveränderungen durch Tumor
Die Ewing-Sarkom-Zellen tragen typischerweise tumorspezifische Genveränderungen, bei denen ein Teil eines Gens auf ein anderes Gen übertragen wird – auch Translokation genannt. Dadurch verändern sich die Aktivierungs- und Signalwege in den Zellen. Die Konsequenz: Die Zellen entarten. Sie werden bösartig und vermehren sich unkontrolliert.
Bei der häufigsten Translokation (85 %) wird ein Stück von Chromosom 22 (EWSR1) mit einem Stück von Chromosom 11 (Fli1) verbunden – auch t(11;22)(q24;q12)-Translokation genannt.
Die Translokationen sind für Ewing-Sarkome so typisch, dass ihr Nachweis zur Diagnose der Erkrankung genutzt wird. Generell werden solche im Tumorgewebe nachweisbaren Genveränderungen nicht vererbt. Das Ewing-Sarkom kann nach Behandlung einer anderen Krebserkrankung als zweite Krebsart auftreten.
Beschwerden des Ewing-Sarkoms
Die Knochenkrebs-Symptome sind sehr unspezifisch. Die Betroffenen leiden in der Regel unter Schmerzen. Anfänglich jedoch nur in Schüben und nicht durchgehend. Bei zunehmendem Tumorwachstum treten die Schmerzen auch nachts auf und es kommt zu Schwellungen der betroffenen Körperregionen. Bewegungseinschränkungen sind ebenfalls ein Symptom.
Oft werden die Knochenkrebs-Schmerzen zunächst fehlinterpretiert – z.B. als Wachstumsschub oder als Folge leichter Verletzungen. Auch die Schwellungen werden häufig zunächst kleinen, vorausgegangenen Verletzungen zugeschrieben. Fieber, Krankheitsgefühl und Gewichtsverlust treten derweil bei nur wenigen Erkrankten auf.
Zwischen den ersten Anzeichen und der Diagnose vergehen oft Wochen bis Monate.

Wichtig: Selbstverständlich hat nicht jedes Kind oder Erwachsener mit den oben beschriebenen Symptomen einen bösartigen Knochenkrebs. Die Erkrankung ist selten, dennoch ist es ratsam, jede Form von Schmerz, die länger als 4 Wochen dauert und nicht eindeutig durch etwas anderes zu erklären ist, ärztlich sorgfältig abklären zu lassen –insbesondere im Kindes- und Jugendalter.
Was tun bei einem Verdacht?
Bei Verdacht auf einen bösartigen Knochentumor sollte ein erfahrenes Zentrum aufgesucht werden: Ein Sarkomzentrum mit Kinderonkologie/Hämatologie und medizinischer Onkologie und einer spezialisierten Sarkomchirurgie und Strahlentherapie.
Unsere Klinik – das Sarkomzentrum des Westdeutschen Tumorzentrums (WTZ) – gehört zu den größten Sarkomzentren in Europa und behandelt deutschlandweit die meisten Menschen mit einer Sarkomerkrankung.
Am Sarkomzentrum des WTZ arbeitet ein hochspezialisiertes Team aus den folgenden Bereichen zusammen:
- Onkologie
- Chirurgie
- Strahlentherapie
- Radiologie
- Nuklearmedizin
- Physiotherapie
- Bewegungstherapie
- Pathologie
Alle Patienten und Patientinnen werden im gesamten Team mehrfach während der Behandlung in Tumorkonferenzen diskutiert, um eine optimale Therapie zu ermöglichen.

Diagnose
Therapie

Nachsorge